Της Δρ. Ανδρούλλας Ν. Μηλιώτου
Η 8η Μαΐου κάθε χρόνου είναι αφιερωμένη στα θαλασσαιμικά σύνδρομα, τα οποία εντάσσονται σε μια συγκεκριμένη κατηγορία κληρονομικών ανωμαλιών παραγωγής αιμοσφαιρίνης, γνωστή ως αιμοσφαιρινοπάθειες. Η αιμοσφαιρινοπάθεια μπορεί να οφείλεται σε ποιοτικές-δομικές ή ποσοτικές μεταβολές του μορίου της αιμοσφαιρίνης. Η πιο κοινή δομικά μη φυσιολογική αιμοσφαιρίνη είναι η HbS. Από την άλλη πλευρά, η ποσοτική παραγωγή των πρωτεϊνών α-/β-σφαιρίνης προκαλεί σύνδρομα θαλασσαιμίας. Αυτά τα μονογονιδιακά σύνδρομα, τα οποία προκαλούνται από ένα μόνο ελαττωματικό γονίδιο, είναι οι πιο διαδεδομένες και κλινικά σημαντικές διαταραχές στον κόσμο.
Η πρώτη κλινική περιγραφή αυτής της κατηγορίας διαταραχών εμφανίζεται το 1928 από τον Cooley(1). Τα σημαντικά αυτά ευρήματα συνοδεύονταν από κλινικές αλλά και εργαστηριακές ενδείξεις μικροκυττάρωσης, υποχρωμίας και κυρίως αυξημένης αντίστασης των ερυθροκυττάρων των ασθενών σε υποτονικά διαλύματα χλωριούχου νατρίου (NaCl). Έκτοτε, οι αιμοσφαιρινοπάθειες αποτελούν σταθερό σημείο αναφοράς καθώς και μοντέλο για τη μελέτη των μονογονιδιακών διαταραχών σε κλινικό, βιοχημικό και μοριακό επίπεδο. Οι μελέτες αυτές οδήγησαν στην κατανόηση της φαινοτυπικής τους ετερογένειας και συνέβαλαν στη διερεύνηση των γνώσεων σχετικά με την παθοφυσιολογία αυτών των μονογονιδιακών διαταραχών, προτείνοντας νέους τρόπους στον τομέα της πρόληψης και της θεραπείας τους. Έχουν περιγραφεί περισσότερες από 1000 μεταλλάξεις στα γονίδια της α- και β-σφαιρίνης, οι οποίες επηρεάζουν τόσο τη δομή όσο και τη σύνθεση των αλυσίδων της αιμοσφαιρίνης.
Οι μέθοδοι κλινικής αντιμετώπισης έχουν βελτιωθεί σημαντικά τα τελευταία χρόνια και το προσδόκιμο ζωής των προσβεβλημένων ατόμων έχει αυξηθεί σημαντικά, με τους ασθενείς να επιβιώνουν μέχρι και την τρίτη με πέμπτη δεκαετία. Για τη θεραπεία των αιμοσφαιρινοπαθειών διατίθεται μόνο υποστηρικτική διαχείριση, δεδομένου ότι η μεταμόσχευση μυελού των οστών, η μόνη αποτελεσματική θεραπεία, είναι πολύ δαπανηρή, αντιμετωπίζει περιορισμούς και δεν αποτελεί ρεαλιστικό μέσο ελέγχου των διαταραχών για πολλές αναπτυσσόμενες χώρες. Ως εκ τούτου, πολλές χώρες, ιδίως εκείνες με υψηλή συχνότητα εμφάνισης θαλασσαιμιών, έχουν εφαρμόσει μια εναλλακτική μέθοδο ελέγχου, η οποία περιλαμβάνει τον έλεγχο του πληθυσμού για φορείς, τον προσδιορισμό του κινδύνου μεταξύ ζευγαριών και την παροχή προγεννητικής διάγνωσης(2).
Ένα από τα κύρια χαρακτηριστικά της γονιδιακής έκφρασης των οικογενειών γονιδίων σφαιρίνης είναι ο αυστηρός έλεγχος της ανισορροπίας και η μειωμένη παραγωγή των προϊόντων του γονιδίου της β-σφαιρίνης, παρατηρείται η εκδήλωση της β-θαλασσαιμίας. Επιδημιολογικά, περίπου το 5% του παγκόσμιου πληθυσμού έχει μεταλλάξεις σε τουλάχιστον ένα αλληλόμορφο των γονιδίων σφαιρίνης, αλλά μόνο το 1,7% πάσχει από α- ή β-θαλασσαιμία.
Η β-θαλασσαιμία αναφέρθηκε για πρώτη φορά στη λεκάνη της Μεσογείου και για το λόγο αυτό πήρε και την ονομασία μεσογειακή αναιμία ή αναιμία Cooley, λόγω της πρώτης κλινικής περιγραφής της το 1928 από τον T.B. Cooley. Φυσικά, δεν είναι τυχαίο ότι η ονομασία β-θαλασσαιμία προέρχεται από τις ελληνικές λέξεις “θάλασσα” (θάλασσα) και “æma” (αίμα). Επιπλέον, φαίνεται να είναι πολύ συχνή σε χώρες που πλήττονται επίσης από την ελονοσία. Η επίπτωση της β-θαλασσαιμίας ποικίλλει ευρέως, ανάλογα με τον εθνικό πληθυσμό. Η νόσος αναφέρεται περισσότερο σε μεσογειακούς, αφρικανικούς και νοτιοανατολικούς ασιατικούς πληθυσμούς. Η υψηλότερη συχνότητα φορέων αναφέρεται στην Κύπρο (14%), τη Σαρδηνία (10,3%) και τη Νοτιοανατολική Ασία(3). Βέβαια, η ανθρώπινη μετανάστευση είχε ως αποτέλεσμα την εμφάνιση της νόσου αυτής σε πολλές άλλες περιοχές του κόσμου(4). Η πληθυσμιακή μετανάστευση και οι μεικτοί γάμοι εισήγαγαν τη μεσογειακή αναιμία σε όλες σχεδόν τις χώρες του κόσμου, συμπεριλαμβανομένης της Βόρειας Ευρώπης, όπου προηγουμένως δεν υπήρχε μεσογειακή αναιμία(87). Περίπου το 1,5% του παγκόσμιου πληθυσμού (80-90 εκατομμύρια) είναι φορείς της β-θαλασσαιμίας, ενώ περίπου 60.000 συμπτωματικά άτομα γεννιούνται ετησίως. Η επίπτωση των συμπτωματικών ατόμων εκτιμάται σε 1 στα 100.000 παγκοσμίως και 1 στα 10.000 στην Ευρώπη(3). Πιο συγκεκριμένα, στην Κύπρο έχει αναφερθεί ότι περίπου ένας (1) στους δέκα (10) Κύπριους είναι φορέας β-θαλασσαιμίας, ενώ σήμερα υπάρχουν περίπου 600 ασθενείς με τη νόσο.
Η β-θαλασσαιμία κληρονομείται ως αυτοσωμική υπολειπόμενη διαταραχή. Υπάρχουν εκατοντάδες μεταλλάξεις εντός του γονιδίου της β-σφαιρίνης, αλλά περίπου 20 διαφορετικά αλληλόμορφα αντιπροσωπεύουν το 80% των μεταλλάξεων που απαντώνται παγκοσμίως. Σε κάθε γεωγραφικό πληθυσμό υπάρχουν μοναδικές μεταλλάξεις. Η πλειονότητα αυτών των μεταλλάξεων είναι σημειακές νουκλεοτιδικές αλλαγές, για παράδειγμα αντικαταστάσεις μιας βάσης, διαγραφές ή εισαγωγές λίγων νουκλεοτιδίων. Οι διαγραφές του γονιδίου της β-σφαιρίνης είναι πολύ σπανιότερες. Σε πληθυσμούς όπου η β-θαλασσαιμία είναι συχνή, η πλειονότητα των μεταλλαγμένων αλληλόμορφων καλύπτεται από λίγες κοινές μεταλλάξεις και τα υπόλοιπα παθολογικά αλληλόμορφα έχουν μεγαλύτερο αριθμό σπάνιων μεταλλάξεων(5).
Όλοι οι ασθενείς παρουσιάζουν ελαττώματα στην παραγωγή της β-αλυσίδας της αιμοσφαιρίνης (HbB), αλλά οι φαινότυποι που προκύπτουν είναι εξαιρετικά ποικίλοι και κυμαίνονται από σοβαρή αναιμία έως απουσία κλινικών συμπτωμάτων. Η ταξινόμηση και ο βαθμός σοβαρότητας της νόσου βασίζεται κυρίως στα επίπεδα αιμοσφαιρίνης, ανεξάρτητα από τον υποκείμενο γονότυπο. Οι τρεις κύριες κατηγορίες στις οποίες ταξινομούνται οι φαινότυποι της β-θαλασσαιμίας είναι (με αύξουσα σειρά βαρύτητας της νόσου):
a. Ελαφρά β-θαλασσαιμία, όπου το άτομο είναι ασυμπτωματικό,
b. Intermedia β-θαλασσαιμία, και η κατηγορία αυτή περιλαμβάνει κάθε φαινότυπο, ο οποίος δεν αντιστοιχεί στις δύο ακραίες κατηγορίες,
c. Μείζων ομοζυγωτική β-θαλασσαιμία ή μεσογειακή αναιμία ή νόσος του Cooley, η οποία αποτελεί τον πιο σοβαρό φαινότυπο, όπου παρατηρείται σοβαρή αναιμία και ο ασθενής εξαρτάται από μετάγγιση(4).
Οι ασθενείς με ενδιάμεση β-θαλασσαιμία έχουν συγκεντρώσεις αιμοσφαιρίνης στο αίμα 7-10 g/dl και δεν χρειάζονται τακτική μετάγγιση. Οι ασθενείς μπορεί να παρουσιάζουν ένα ευρύ φάσμα κλινικών συμπτωμάτων, ανάλογα με τον βαθμό ανισορροπίας μεταξύ α- και β-σφαιρίνης και διάφορους γενετικούς και περιβαλλοντικούς παράγοντες. Οι ασθενείς μπορεί να υποφέρουν από πολλές επιπλοκές, όπως πνευμονική υπέρταση, θρομβωτικά συμβάντα, λοίμωξη, ενδοκρινική δυσλειτουργία και έλκη στα πόδια(6).
Οι ασθενείς με μείζονα β-θαλασσαιμία χρειάζονται τακτικές μεταγγίσεις ερυθρών αιμοσφαιρίων για να επιβιώσουν(7). Ωστόσο, οι επαναλαμβανόμενες μεταγγίσεις προκαλούν υπερφόρτωση σιδήρου, με απειλητικές για τη ζωή επιπλοκές όπως ενδοκρινική δυσλειτουργία, καρδιομυοπάθεια, ηπατική νόσο και τελικά πρόωρο θάνατο. Ελλείψει μετάγγισης, οι ασθενείς με μείζονα β-θαλασσαιμία πεθαίνουν εντός των πρώτων πέντε ετών της ζωής τους, και ακόμη και με μεταγγίσεις, μόνο το 50-65% των ασθενών ζουν πέραν της ηλικίας των 35 ετών σε χώρες υψηλού εισοδήματος(8). Η έρευνα γύρω από τη β-θαλασσαιμία είναι πολύ ενεργή και έχει σημειωθεί πρόοδος στην ανάπτυξη νέων θεραπευτικών προσεγγίσεων, ορισμένες από τις οποίες βρίσκονται σε στάδιο κλινικών δοκιμών(9).
Η γονιδιακή θεραπεία είναι ένα πολλά υποσχόμενο “εφάπαξ φάρμακο”, χωρίς την ανάγκη (θεωρητικά) για ανοσοκατασταλτικά σχήματα και προφύλαξη από την νόσο του μοσχεύματος-έναντι-ξενιστή (Graft-versus-host disease, GVHD).
Η γονιδιακή θεραπεία για τη β-θαλασσαιμία απαιτεί τη διαμόλυνση του γονιδίου σε αιμοποιητικά βλαστοκύτταρα (HSCs), χρησιμοποιώντας φορείς που κατευθύνουν τη ρυθμιζόμενη έκφραση της β-σφαιρίνης σε θεραπευτικά επίπεδα(10). Για παράδειγμα, η προσέγγιση της γονιδιακής θεραπείας, με τη χρήση ενός ρετροϊικού φορέα που φέρει το γονίδιο της ανθρώπινης β-σφαιρίνης, έλαβε πρόσφατα έγκριση υπό όρους από τον FDA και τον Ευρωπαϊκό Οργανισμό Φαρμάκων (EMA) για τη θεραπεία της εξαρτώμενης από τη μετάγγιση μεσογειακής αναιμίας (TDT) και είναι η μόνη θεραπεία υπό αξιολόγηση και με δεσμεύσεις μετά την κυκλοφορία στην αγορά. Πιο συγκεκριμένα, ο EMA έδωσε άδεια κυκλοφορίας υπό όρους (Conditional Marketing Authorization/CMA) για το αυτοτελές φάρμακο betibeglogene autotemcel (beti-cel) (εμπορική ονομασία: Zynteglo), η οποία τελικά ανεστάλη, λόγω του εξαιρετικά υψηλού κόστους. Ο FDA έδωσε άδεια κυκλοφορίας για το Zynteglo το 2022 και είναι η πιο ακριβή θεραπεία (2,8 εκατ. δολάρια) που έχει κυκλοφορήσει ποτέ στις ΗΠΑ.
Προς αυτή την κατεύθυνση των βιοτεχνολογικά ανασυνδυασμένων πρωτεϊνών, η ομάδα μας από το Εργαστήριο Φαρμακολογίας, του Τμήματος Φαρμακευτικής, του Αριστοτελείου Πανεπιστημίου Θεσσαλονίκης έχει πραγματοποιήσει εκτεταμένη εργασία για τις αιμοσφαιρινοπάθειες. Πιο συγκεκριμένα, προτείναμε την παραγωγή ανασυνδυασμένης αλυσίδας α- και β-σφαιρίνης, συντηγμένης με το πεπτίδιο TAT, ως πρωτεϊνική θεραπευτική προσέγγιση των α- και β- θαλασσαιμιών. Η ενδοκυτταρική μεταγωγή της βιοτεχνολογικά ανασυνδυασμένης πρωτεΐνης TAT-β-σφαιρίνης-HA καταδείχθηκε με επιτυχία σε ανθρώπινα κύτταρα ερυθρολευχαιμίας K-562. Στα κύτταρα αυτά υπάρχει αδυναμία παραγωγής της ενήλικης HbA (α2β2), λόγω της αδυναμίας τους να παράγουν την ώριμη αλυσίδα β-σφαιρίνης [και για το λόγο αυτό παράγονται σε αυξημένα επίπεδα οι εμβρυϊκές Hb Gower-1 (ζ2ε2), Hb Portland (ζ2γ2), καθώς και η Hb Gower-2 (α2ε2)] και η εμβρυϊκή HbF (α2γ2), μετά από έκθεση σε αιμίνη. Η παραγόμενη ΤΑΤ-β-σφαιρίνη-ΗΑ έχει τη δυνατότητα ενδοκυττάριας μεταγωγής και συνεπώς αντικατάστασης των ελλείπουσων ενδογενών β-σφαιρινών, ενώ διαπιστώθηκε επίσης η αλληλεπίδραση με τις ενδογενείς α-αλυσίδες και ο σχηματισμός α2β2-τετραμερών, αλλά σε περιορισμένο βαθμό(11).
Το 2021, η ομάδα μας, Miliotou, et al., πρότεινε επίσης την ex vivo PTD-μεσολαβούμενη χορήγηση αλυσίδας α-σφαιρίνης σε ερυθροκύτταρα ασθενών με α-θαλασσαιμία (HbH) στο πλαίσιο της θεραπείας πρωτεϊνικής αντικατάστασης. Οι ανθρώπινες ανασυνδυασμένες πρωτεΐνες σύντηξης α-σφαιρίνης-ΗΑ καθώς και η ΤΑΤ-α-σφαιρίνη-ΗΑ δημιουργήθηκαν μέσω της κλωνοποίησης της κωδικής αλληλουχίας της α-σφαιρίνης, συγχωνευμένης με την νουκλεοτιδική αλληλουχία του πεπτιδίου ΤΑΤ, στην τελευταία. Η χρωματογραφία αποκλεισμού μεγέθους χρησιμοποιήθηκε για την αξιολόγηση και την επιβεβαίωση της ικανότητας της ανθρώπινης ανασυνδυασμένης α-σφαιρίνης-HA να αλληλεπιδρά in vitro με την προηγουμένως συντιθέμενη ΤΑΤ-β-σφαιρίνη-HA και να δημιουργεί ετεροδιμερή α-/β-σφαιρίνης. Κατά τη διάρκεια των αρχικών μελετών αξιολόγησης της μεταγωγής, η ανασυνδυασμένη ΤΑΤ-α-σφαιρίνη-HA μεταφέρθηκε επιτυχώς σε ανθρώπινα κύτταρα Κ-562. Τέλος, τα HbH-Inclusion Bodies, τα οποία είναι γνωστό ότι περιλαμβάνουν τοξικά τετραμερή της αλυσίδας α-σφαιρίνης, μειώθηκαν με τη μεταγωγή ανασυνδυασμένης ΤΑΤ-α-σφαιρίνης-ΗΑ σε ερυθροκύτταρα ασθενών με HbH(12).
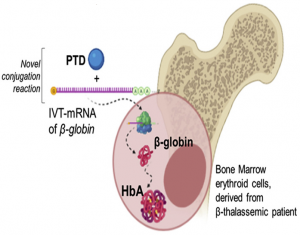
Τα τελευταία χρόνια, μια εναλλακτική προσέγγιση της θεραπείας πρωτεϊνικής αντικατάστασης είναι το in vitro μεταγραφόμενο (IVT)-mRNA, το οποίο έχει προσελκύσει την προσοχή της επιστημονικής κοινότητας και αποτελεί έναν από τους πιο ισχυρούς και πολλά υποσχόμενους υποψηφίους για την ανάπτυξη καινοτόμων θεραπευτικών προσεγγίσεων για τις γενετικές διαταραχές, ενώ ο κατάλογος των εφαρμογών του συνεχίζει να αυξάνεται. Μια σημαντική πρόκληση για την επιτυχή εφαρμογή της τεχνολογίας IVT-mRNA είναι η ανάπτυξη μιας ασφαλούς και αποτελεσματικής μεθόδου χορήγησης. Στην πρόσφατη μελέτη μας, αναπτύξαμε μια νέα μέθοδο για την παροχή μιας ασφαλούς πλατφόρμας μεταφοράς, χρησιμοποιώντας την τεχνολογία πεπτιδίων μεταγωγής (Protein Transduction Domain, PTD) για την ενδοκυτταρική μεταγωγή θεραπευτικών IVT-mRNA και την επακόλουθη έκφραση των επιθυμητών πρωτεϊνών. Εν συντομία, η ομάδα μας παρουσίασε μια νέα πλατφόρμα παράδοσης χρησιμοποιώντας οποιοδήποτε δυνητικά θεραπευτικό (ή μη) IVT-mRNA, συζευγμένο με ένα υδρόφοβο πεπτίδιο μεταγωγής, μήκους 6 αμινοξέων, το PFVYLI. Η μοναδική ομοιοπολική χημική αντίδραση, που συνδέει το PFVYLI με οποιοδήποτε IVT-mRNA κατοχυρώθηκε με δίπλωμα ευρεσιτεχνίας ως μεθοδολογία για τη δημιουργία μιας καθολικής πλατφόρμας ενδοκυττάριας μεταφοράς, χρησιμοποιώντας την πουρομυκίνη, μέσω ενός αμιδικού δεσμού, που δρα ως συνδετικό στοιχείο με το IVT-mRNA. Η διεθνής αίτηση διπλώματος ευρεσιτεχνίας δημοσιεύθηκε με τον αριθμό WO2021/094792A1 και τίτλο: «Method for the development of a delivery platform to produce deliverable PTD-IVT-mRNA therapeutics» (13). Ο Εθνικός Οργανισμός Βιομηχανικής Ιδιοκτησίας (ΟΒΙ) χορήγησε στην ομάδα μας ελληνικό δίπλωμα ευρεσιτεχνίας με αριθμό 1010063, το οποίο ισχύει έως το 2039. Παράλληλα, υποβλήθηκε αίτηση στο Ευρωπαϊκό Γραφείο Διπλωμάτων Ευρεσιτεχνίας με τον αριθμό P20823912.9.
Το PTD-IVT-mRNA της ανθρώπινης β-σφαιρίνης παρουσίασε αποτελεσματική διαμόλυνση του IVT-mRNA και επιτυχή, ανθεκτική έκφραση πρωτεΐνης σε κύτταρα K-562, ως μοντέλο κυτταρικής σειράς (13). Η κινητική της ενδοκυτταρικής μεταφοράς σε επίπεδο mRNA, καθώς και της πρωτεϊνικής μετάφρασης, που προκαλείται από την ενδοκυτταρική μεταγωγή του PTD-IVT-mRNA της ανθρώπινης β-σφαιρίνης, μελετήθηκε για διάφορα χρονικά διαστήματα έως και 96 ώρες και από τα πρώτα 30 λεπτά της επώασης των κυττάρων, με αυξημένη πρωτεϊνική έκφραση για όλα τα χρονικά σημεία που εξετάστηκαν μετά την επώαση. Στο χρονικό σημείο των 72 ωρών, τα επίπεδα έκφρασης της β-σφαιρίνης τριπλασιάστηκαν στατιστικά σημαντικά, από το αντίστοιχο ποσοστό στα κύτταρα που δεν είχαν επωαστεί. Επιπλέον, τα PTD-IVT-mRNAs δεν είχαν καμία επίδραση στην κυτταρική βιωσιμότητα και ανάπτυξη, τόσο σε αιωρούμενα κύτταρα όσο και σε προσκολλημένα κύτταρα, ενώ δεν παρατηρήθηκε καμία τοξικότητα και καμία φαινοτυπική αλλαγή στα επεξεργασμένα κύτταρα.
Τέλος, η αξιολόγηση του PTD-IVT-mRNA της ανθρώπινης β-σφαιρίνης ολοκληρώθηκε με επιτυχία σε κύτταρα του μυελού των οστών, που προέρχονταν από τρεις διαφορετικούς β-θαλασσαιμικούς ασθενείς. Στα πειράματα που πραγματοποιήθηκαν, ομοίως, η ενδοκυτταρική μεταγωγή του PTD-IVT-mRNA της ανθρώπινης β-σφαιρίνης στα κύτταρα του μυελού των οστών των ασθενών επιβεβαιώθηκε από τα 30 λεπτά επώασης και παρέμεινε αυξημένη έως και 96 ώρες, όπως ελέγχθηκε σε δύο από τους τρεις ασθενείς. Συνολικά, και για τους τρεις ασθενείς παρατηρήθηκε ότι η αύξηση των ενδοκυττάριων ποσοστών έκφρασης της β-σφαιρίνης ήταν στατιστικά σημαντικά μεγαλύτερη, σε σχέση με τα μη επεξεργασμένα κύτταρα (13). Η νέα μας πλατφόρμα PTD-IVT-mRNA σχεδιάστηκε για να προσφέρει πολυάριθμες βελτιώσεις, σε σχέση με τις προηγούμενες πλατφόρμες ενδοκυττάριας μεταφοράς, καθώς αντιμετωπίζει με επιτυχία τα προβλήματα σταθερότητας, μεταγωγής και μετάφρασης στον τομέα των IVT-mRNA-θεραπευτικών.
Συμπερασματικά, η τεχνολογία PTD έχει επιστρατευθεί εκτενώς από την ερευνητική μας ομάδα για την αντιμετώπιση της θαλασσαιμίας. Εφαρμόστηκε με επιτυχία in vitro και ex vivo ενδοκυττάρια μεταφορά ανασυνδυασμένων α- και β-σφαιρινών σε μοντέλα αιμοσφαιρινοπαθειών, όπως η α- και β- θαλασσαιμία. Επεκτείνοντας τους ορίζοντες, η ομάδα μας έκανε ένα βήμα μπροστά και προσέγγισε αυτές τις διαταραχές, χρησιμοποιώντας το αντίστοιχο IVT-mRNA του γονιδίου της β-σφαιρίνης και εφάρμοσε με επιτυχία την θεραπεία πρωτεϊνικής αντικατάστασης και πάλι χρησιμοποιώντας την τεχνολογία PTD, μέσω μιας καινοτόμου, κατοχυρωμένης με δίπλωμα ευρεσιτεχνίας αντίδρασης σύζευξης με το IVT-mRNA.
Έτσι, η θεραπεία μονογονιδιακών διαταραχών, όπως οι θαλασσαιμίες, μπορεί είναι εφικτό να προσεγγιστεί με πολλούς τρόπους και τα ευρήματά μας παρέχουν μια πολύπλευρη προοπτική, εφαρμόζοντας αποτελεσματικά είτε ανασυνδυασμένες πρωτεΐνες είτε θεραπευτικού IVT-mRNA για την αντιμετώπιση των θαλασσαιμικών συνδρόμων.
(1) COOLEY TB. Obituaries. Am J Dis Child. 1946;71(1):77-9
(2) Peng CT, Liu SC, Peng YC, Lin TH, Wang SJ, Le CY, et al. Distribution of thalassemias and associated hemoglobinopathies identified by prenatal diagnosis in Taiwan. Blood cells, molecules & diseases. 2013;51(3):138-41 10.1016/j.bcmd.2013.04.007.
(3) Hassan T, Badr M, El Safy U, Hesham M, Sherief L, Zakaria M. β-Thalassemia: Genotypes and Phenotypes. Epidemiology of Communicable and Non-Communicable Diseases – Attributes of Lifestyle and Nature on Humankind. IntechOpen2016.
(4) Flint J, Harding RM, Boyce AJ, Clegg JB. The population genetics of the haemoglobinopathies. Bailliere’s clinical haematology. 1998;11(1):1-51 10.1016/s0950-3536(98)80069-3.
(5) Thein SL. Molecular basis of beta thalassemia and potential therapeutic targets. Blood cells, molecules & diseases. 2018;70:54-65 10.1016/j.bcmd.2017.06.001.
(6) Borgna-Pignatti C, Marsella M, Zanforlin N. The natural history of thalassemia intermedia. Annals of the New York Academy of Sciences. 2010;1202:214-20 10.1111/j.1749-6632.2010.05550.x.
(7) Rachmilewitz EA, Giardina PJ. How I treat thalassemia. Blood. 2011;118(13):3479-88 10.1182/blood-2010-08-300335.
(8) Borgna-Pignatti C. The life of patients with thalassemia major. Haematologica. 2010;95(3):345-8 10.3324/haematol.2009.017228.
(9) de Dreuzy E, Bhukhai K, Leboulch P, Payen E. Current and future alternative therapies for beta-thalassemia major. Biomedical journal. 2016;39(1):24-38 10.1016/j.bj.2015.10.001.
(10) Gaziev J, Lucarelli G. Allogeneic cellular gene therapy for hemoglobinopathies. Hematology/oncology clinics of North America. 2010;24(6):1145-63 10.1016/j.hoc.2010.08.004.
(11) Papadopoulou LC, Ingendoh-Tsakmakidis A, Mpoutoureli CN, Tzikalou LD, Spyridou ED, Gavriilidis GI, et al. Production and Transduction of a Human Recombinant beta-Globin Chain into Proerythroid K-562 Cells To Replace Missing Endogenous beta-Globin. Molecular pharmaceutics. 2018;15(12):5665-77 10.1021/acs.molpharmaceut.8b00857.
(12) Miliotou AN, Papagiannopoulou D, Vlachaki E, Samiotaki M, Laspa D, Theodoridou S, et al. PTD-mediated delivery of α-globin chain into Κ-562 erythroleukemia cells and α-thalassemic (HBH) patients’ RBCs ex vivo in the frame of Protein Replacement Therapy. Journal of Biological Research-Thessaloniki. 2021;28(1):16 10.1186/s40709-021-00148-3.
(13) Miliotou AN, Pappas IS, Spyroulias GA, Vlachaki E, Tsiftsoglou AS, Vizirianakis IS, et al. Development of a novel PTD-mediated IVT-mRNA delivery platform for potential protein replacement therapy of metabolic/genetic disorders. Molecular Therapy – Nucleic Acids. 2021;26: 694-710 https://doi.org/10.1016/j.omtn.2021.09.008.